
Spermidine ameliorates the neuronal aging by improving the mitochondrial function in vitro
Changes in mitochondrial structure and function are the initial factors of cell aging. Spermidine has an antiaging effect, but its effect on neuronal aging and mitochondrial mechanisms is unclear. In this study, mouse neuroblastoma (N2a) cells were treated with D‑galactose (D‑Gal) to establish cell aging to investigate the antiaging effect and mechanisms of spermidine. Changes in the cell cycle and β-galactosidase activity were analyzed to evaluate the extent of cell aging. Stabilities of mitochondrial mRNA and mitochondrial membrane potential (MMP) were evaluated in the process of cell aging under different treatments. The mitochondrial function was also evaluated using the Seahorse Metabolic Analysis System combined with ATP production. The unfolded protein response (UPR) of the N2a cells was analyzed under different treatments. Results showed that spermidine pretreatment could delay the cell aging and could maintain the mitochondrial stability during D‑Gal treatment. Spermidine increased the proportion of cells in the S phase and maintained the MMP. The oxygen utilization and ATP production in the N2a cells were reduced by D‑Gal treatment but were partially rescued by the spermidine pretreatment. Spermidine ameliorated the N2a cell aging by promoting the autophagy and inhibiting the apoptosis except the UPR. These results showed that spermidine could ameliorate the N2a cell aging by maintaining the mitochondrial mRNA transcription, MMP and oxygen utilization during the D‑Gal treatment.
Aging is the most important risk factor for neurodegenerative diseases. Neurodegenerative diseases, such as Parkinson disease (PD) and Alzheimer's disease (AD), exhibit typical age-dependent characteristics (de Lau et al., 2004; Rubinsztein and Easton, 1999). This point suggests that the factors accelerating aging are also involved in the development of neurodegenerative diseases. The mechanisms of aging are complex and many factors were involved, such as genomic stability decline, epigenetic change, nutritional disorders, proteolysis abnormality, telomere shortening, stem cell depletion and mitochondrial dysfunction (Gems and Partridge, 2013; Kenyon, 2010; Lopez-Otin et al., 2013). Among of them, mitochondrial function decline is a critical factor in neuronal aging, which is attributed to the high oxygen consumption in the process of neuron metabolism (Lopez-Otin et al., 2016; Macedo et al., 2017). Recently, mitochondrial dysfunction was found to be involved in the development of brain aging (Basha and Poojary, 2014), especially mitochondrial DNA (mtDNA) mutation and fragment deletion, which existed in the early times (Lauri et al., 2014). In one study, mice with abnormal mtDNA repair exhibited a typical aging phenotype in the postnatal period for two months and shortening of whole life span (Trifunovic et al., 2004). The protective effect of free radical scavengers on the mitochondria is also the main mechanism of antiaging and in reducing neurodegenerative diseases (Sano et al., 1997; Zhao et al., 2008)
Spermidine is a trivalent cationic compound containing amino groups in eukaryotic cells. It is synthesized by butanediamine and Sadenosylmethionine (Gosule and Schellman, 1976). Under normal conditions, spermidine is alkaline and exists as protonated forms under physiological conditions. Spermidine can interact with nucleic acids, proteins, ATP and other polyanions through electrostatic binding, and is involved in multiple functions, such as maintenance of DNA genomic homeostasis, gene transcription and translation regulation, regulation of cell proliferation, and maintenance of cell survival (Childs et al., 2003; Pegg, 1988). In the central nervous system, polyamines (spermidine and spermine) accumulate preferentially in glial cells but not in neurons (Biedermann et al., 1998; Laube and Veh, 1997). By contrast, Krauss et al. reported that polyamine synthesis is absent in glia cells but its heterogeneous expression predominantly localized to neurons and neuropil (Krauss et al., 2007). This phenomenon suggests that brain obtains polyamines from external sources; but inside the brain, the major sources of polyamines are the glial cells that store them (Skatchkov et al., 2000; Skatchkov et al., 2014). Spermidine exerts a protective effect on the neuronal oxidative stress, inflammation and local ischemia injury through the inhibition of histone acetyltransferase activity, reduction of histone-3 acetylation and regulation of specific gene expression (Minois et al., 2012).
Spermidine regulates cell growth, differentiation and death and also stabilizes DNA and RNA structures and various biological membranes (Wang et al., 2009) . Spermidine is an antioxidant, nutrient and the second messenger in cells (Khomutov et al., 2009). It can induce cell autophagy and prolong the life spans of yeast, fruit fly, nematode and human immune cells (Eisenberg et al., 2009; Soda et al., 2009). Recently, Bell et al. reported that the protective effect of pentylenetetrazole in a model of epilepsy depends on the increase of putrescine which is the simplest polyamine, then the putrescine converted into the GABA in the presynaptic neurons. This results demonstrated polyamine has unknown roles in the development brain. (Bell et al., 2011). In another study, Noro et al. reported that spermidine promoted retinal ganglion cell survival and optic nerve regeneration by inhibiting the active microglia and inflammation (Noro et al., 2015). These evidences suggested Potential roles of polyamine are complex in nervous system and need to be investigated. Spermidine can ameliorate the damage from oxidative stress in aging mice and upregulate the autophagy activity through chromatin acetylation to anti-aging in yeast, fruit fly, nematodes, and human cells (Minois, 2014).
Target elimination of abnormal mitochondria depends on autophagy activity. Nevertheless, little is known whether spermidine can maintain the mitochondrial stability under stress conditions through mitochondrial quality control, thereby delaying the neuronal aging. The aim of the present study is to determine the effect of spermidine on neuronal aging and its protective mechanisms.
2. Materials and methods
Galactose (D‑Gal, catalog no. G0625), D‑glucose (catalog no. 552003), D‑mannitol (catalo no. 240184), spermidine (catalog no. 85561), 3‑4,5‑dimethyl‑2‑thiazolyl‑2,5‑diphenyl‑2H‑tetrazolium bromide (MTT, catalog no. M5655) and Rhodamine 123 (Rh123, catalog no. R8004) were purchased from Sigma (St. Louis, MO, USA); The βgalactosidase staining kit and protein extraction kit were purchased from Beyotime (Beyotime Biotechnology Co., Ltd., Shanghai, China; catalog no. c0602); The ATP assay kit was purchased from Jiancheng Biotechnology (Jiancheng Biological Company, Nanjing, China; catalog no. A095-1). The oxygen consumption rate (OCR) assay kit was purchased from Agilent (Agilent Co., Ltd., IL, US; catalog no. 103020-100). The calf serum was purchased from Sijiqing Biotechnology (Sijiqing Biotechnology Company, Hangzhou, China). The polyclonal anti-rabbit P53 antibodies were purchased from Abcam (Abcam, Cambridge, UK; catalog no. ab131442). The polyclonal anti-rabbit cleaved caspase-3 (catalog no. Asp175), AMPK (catalog no. 2532) and phosphorylation AMPK antibodies (catalog no. 2531S) were purchased from cell signaling (CST, MA, USA). The polyclonal anti-rabbit LC3 antibody was purchased from Santa Cruz (Santa Cruz, IL, USA; catalog no. sc292354); The monoclonal anti-mouse GAPDH antibody was purchased from Millipore (Millipore, CA, USA; catalog no. AB2302).
The RNAiso Plus kit (catalog no. 9108), PrimeScript RT Master Mix kit and SYBR Premix Ex Taq II kit (catalog no. RR036A) were purchased from TAKARA (TAKARA Biotechnology Co., ltd., Dalian, China). The primers were designed and synthesized by TAKARA (TAKARA Biotechnology Co., ltd., Dalian, China).The mouse neuroblastoma cell (N2a) line was purchased from the Shanghai Cell Institute of the Chinese Academy of Sciences. The complete medium was composed of DMEM high glucose medium and 10% calf serum; the pH value was 7.4; the cell growth in the logarithmic phase was cultured in a 96-well plate; and the cell density was adjusted to 1.5 × 104 /well. After adherence, cells were incubated with at 5, 10, 20 and 30 μM spermidine. After 1 h, the original culture solution was sucked, and the cells were gently washed with the DMEM complete medium twice. Then, the cells were incubated with 50, 100, 200, and 300 mM D‑Gal for 24 h and 48 h as described in previous work (Delwing-de Lima et al., 2017; Li et al., 2014; Xing et al., 2006; Zhang et al., 2015). In brief, D‑Gal, D‑glucose, and D‑mannitol were diluted directly in DMEM and sterilized using a bacterial filter. The volume of solution was modified according to the final concentration. The cell viability was measured using the MTT assay. In brief, cells were incubated with 450 μM MTT for 3 h and then centrifuged at 1800 rpm for 10 min at room temperature to remove the supernatant. Afterwards, formazan was extracted from pelleted cells with 600 μl of DMSO for 15 min.
The amount of MTT-formazan was determined by 570 nm absorbance with 655 nm as the wavelength reference. To evaluate the effects of 100 mM spermidine on osmosis of N2a cells, N2a cells were incubated with the 100 mM D‑glucose and D‑mannitol for 48 h and then the cell viability was measured using MTT assay.N2a cells were treated with 20 μM spermidine. After 1 h, spermidine was washed off, and the cells were incubated with the 100 mM D‑Gal for 48 h. In the control groups, N2a cells were incubated with 100 mM D‑Gal, 100 mM D‑glucose, and 100 mM D‑mannitol for 48 h, repectively, but without the spermidine-pretreatment. The original culture medium was removed, and the cells were gently rinsed twice at 37 °C with 0.01 M PBS (pH, 7.4). A total of 200 μl of fixed solution per well was added, and the mixture was incubated at room temperature for 15 min. The fixed solution was removed and rinsed with 0.01 M PBS for 5 min. Another 200 μl of β-galactosidase staining solution per well was added, and the mixtures was incubated overnight. After removing the staining solution and rinsing with 0.01 M PBS, the number of positive cells was counted in nine areas of view per well under an inverted microscope.N2a cells were treated with 5, 10, 20 and 30 μM spermidine. After 1 h, spermidine was washed off, and 100 mM of D‑Gal was added. The cells were incubated for 48 h, harvested, fixed with 70% ethanol, stained with propidium iodide and detected using flow cytometry. The percentages of cells in the S, G1, and G2 phase were calculated. N2a cells were incubated with 5, 10, and 20 μM spermidine. After 1 h, the spermidine was washed off, and 100 mM of D‑Gal was added. The cells were incubated for 48 h and then harvested for ATP determination.
The operation was performed according to the kit instruction. The absorbance of each well was detected using a microplate reader (exciting light of 636 nm). ATP concentration was calculated according to the following formula: ATP (μmol/g protein) = (OD value of the treated group − OD value of the control group) × sample protein concentration (g prot/l) / (OD value of the standard sample − OD value of the blank well) × standard sample concentration (1 × 103 μmol/l) × dilution ratio.
Mitochondrial membrane potential (MMP) analysis
N2a cells were incubated with 5, 10, and 20 μM spermidine. After 1 h, spermidine was washed off, and 100 mM of D‑Gal was added. The cells were incubated for 48 h and then were harvested for the MMP analysis. Rh123 storage solution was diluted to 1.5 μg/ml concentration with 0.01 M PBS (pH 7.4) as the work solution. The cells were rinsed with 1 ml of 0.01 M cooling PBS. A total of 500 μl of Rh123 working solution was added and developed for 30 min at 37 °C in the dark. Then, the cells were centrifuged at 800 rpm for 10 min and washed with 0.01 M ice cold PBS twice. The cells were suspended with 0.01 M PBS and measured using flow cytometry .
OCR assay
The OCR assay was performed using a Clark-type electrode (Agilent, Seahorse XF24 analyzer) in accordance with previous methods (Yao et al., 2009).
In brief, the N2a cells were incubated with 20 μM spermidine. After 1 h, the spermidine was washed off, and 100 mM of D‑Gal was added. The cells were cultured for 48 h. On the day of the metabolic flux analysis, The medium was changed to unbuffered DMEM (DMEM base medium supplemented with 25 mM glucose, 1 mM sodium pyruvate, 31 mM NaCl, and 2 mM GlutaMax; pH 7.4), and the cells were incubated at 37 °C in a non-CO2 incubator for 1 h. All medium and injection reagents were adjusted to pH 7.4 on the day of assay. Four baseline measurements of OCR were obtained before the sequential injection of mitochondrial inhibitors. Three readings were obtained after each addition of mitochondrial inhibitor before the injection of the subsequent inhibitors. The mitochondrial inhibitors of oligomycin (1 μM), FCCP (1 μM), and rotenone (1 μM) were used. OCR was automatically calculated and recorded using the Seahorse XF-24 software. After the assays, the plates were saved, and protein readings were measured for each well to confirm equal cell numbers per well. In comparison with the basal rates, the percentage of change was calculated as the value of change divided by the average value of baseline readings.
Qualitative RT-PCR
N2a cells were incubated with 5, 10, and 20 μM spermidine. After 1 h, spermidine was washed off, and 100 mM of D‑Gal was added. The cells were incubated for 48 h and then were harvested.
N2a cells were incubated with 5, 10, and 20 μM spermidine. After 1 h, spermidine was washed off, and 100 mM of D‑Gal was added. The cells were incubated for 48 h and then were harvested. The total RNA was extracted using RNAiso Plus and reversely transcribed into cDNA using the PrimeScript RT Master Mix kit. The relative expression levels of the target and reference genes were detected using SYBR Premix Ex Taq II kit through real-time fluorescent quantitative PCR (Thermo, PikoReal 96, USA).
Western blot
N2a cells were incubated with 5, 10, and 20 μM spermidine. After 1 h, spermidine was washed off, and 100 mM or 200 mM of D‑Gal was added. The cells were incubated for 24 h or 48 h and then were harvested. The total protein was extracted using the total protein extraction kit. The protein content was measured using the Bradfold method. The loading buffer was added in the extracted samples, and the samples were boiled and denatured. The loading amount of total protein was maintained to 50 μg. The sample was separated using 10%–12% SDSPAGE, transferred on the PVDF membrane, and blocked with 5% nonfat milk. The cleaved caspase-3 antibody (1:1000), p53 antibody (1:500), AMPK and phosphorylation AMPK antibody (1:1000), and GAPDH antibody (1:1000) were added, and the sample was incubated overnight at 4 °C. The membrane was washed with 0.01 M TBST. The corresponding secondary antibody (1:5000) was added and incubated at room temperature for 2 h. Enhanced chemiluminescence was used for development, and the relative expression was calculated using the bioanalytical imaging system (Azure Biosystem, INC., USA). The target molecular expression was quantified based on the loading control with the GAPDH expression.
Statistical analysis
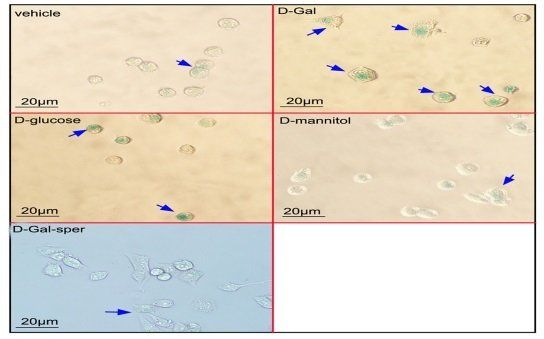
All data are expressed as the mean ± SD of three independent measurements. Statistical analysis was performed using SPSS Statistics software 17.0. Differences between the two groups were analyzed using Student's t-test, whereas those among three or more groups were analyzed using one-way analysis of variance with least significant difference test. Statistical significance was considered at p < 0.05. 3. Results 3.1. Antiaging effect of spermidine on N2a cells following D‑Gal treatment The N2a cells viability was measured following treatment with a different dose of D‑Gal for 24 h or 48 h. The cell viability decreased approximately 25% after 100 mM D‑Gal treatment for 48 h compared with the control (Fig. 1A). The protective effects of spermidine on N2a cell damage induced by D‑gal were tested using the MTT assay. Results showed that the pretreatment with spermidine at 10, 20, and 30 μM for 1 h improved the cell survival following 100 mM D‑Gal treatment (Fig. 1B). The effects of spermidine on D‑Gal induced cell aging were detected using β‑galactosidase staining. After the N2a cells were treated with 100 mM of D‑Gal for 48 h, the number of β‑galactosidase stainingpositive cells increased significantly compared with the control group (p < 0.01).
The spermidine pretreatment reduced the number of β‑galactosidase staining-positive cells compared with the single D‑Gal treatment group (p < 0.01). This result suggested that spermidine ameliorated the D‑Gal induced cell aging (Fig. 1C and D). To exclude the effect of D‑Gal on cell osmosis, N2a cells were incubated with 100 mM D‑glucose and 100 mM D‑mannitol for 48 h, respectively. The cell aging was evaluated using the β-galactosidase staining. And result exhibited ration of positive cells in D‑glucose and D‑mannitol treated groups was same as in the control group (Fig. 1C, and D). The cell viability assay showed D‑Gal-treatment decreased cell viability compared with the D‑glucose or D‑mannitol-treatmen Effect of spermidine on N2a cell cycle following D‑Gal treatment The effects of spermidine on the N2a cell cycle following treatment with 100 mM D‑Gal for 48 h were analyzed using flow cytometry. Results (Fig. 2A and B) showed that the N2a cells treated with 100 mM D‑Gal for 48 h significantly decreased the proportion of cells in the S phase and significantly increased the proportion of cells in the G1 phase compared with the control group (p < 0.01). The 10, 20, and 30 μM spermidine pretreatment increased the proportion of cells in the S phase and decreased the proportion of cells in the G1 phase in a dose-dependent manner. This result suggested that spermidine improved the cell cycle arrest induced by D‑Gal. 3.3. Protective effect of spermidine on mitochondrial damage of N2a cells induced by D‑Gal The effects of spermidine on the mitochondrial genomic stability were evaluated by detecting the mitochondrial mRNA level following the treatment with the 100 mM D‑Gal for 48 h.
that the 100 mM D‑Gal treatment for 48 h significantly decreased the levels of mitochondrial mRNA in the N2a cells compared with the control group (p < 0.01). The 20 μM spermidine pretreatment for 1 h significantly increased the expressions levels of cy1, Cy3, Atp6, p41, P5 and P6 compared with the single D‑Gal treatment groups (p < 0.01, or p < 0.05). This outcome suggested that D‑Gal treatment caused mitochondrial genome damage and the spermidine treatment exerted a protective effect on the mitochondrial genome stability. The MMP was detected using Rh123 staining. The 100 mM D‑Gal treated for 48 h significantly decreased the MMP of the N2a cells compared with the control group (p < 0.05). The spermidine pretreatment for 1 h improved the MMP in a dose-dependent manner (Fig. 3B). To further demonstrate the mitochondrial damage effect on the cell survival, the expression of cleaved caspase-3 was detected. The 100 mM D‑Gal treatment for 48 h significantly increased the cleaved caspase-3 expression level in the N2a cells (Fig. 3C and E). The 5, 10, and 20 μM spermidine pretreatment for 1 h exerted an inhibitory effect on the 100 mM D‑Gal-induced cleaved caspase-3 expression. The 100 mM of D‑Gal treatment for 48 h significantly increased the p53 level; pretreatment with 5, 10, and 20 μM spermidine for 1 h provided an inhibitory effect on 100 mM D‑Gal-induced p53 expression (Fig. 3D and F). 3.4. Effect of spermidine on mitochondrial function of N2a cells following D‑Gal treatment The effects of spermidine on mitochondrial respiration were evaluated by detecting the OCR following treatment with the 100 mM D‑Gal for 48 h.