
Spermidine boosts autophagy to protect from synapse aging
The first kiss, the smell of your family’s cooking or a child’s first word: memories such as these are imprinted tightly into our brains and can be recollected even years later. However, the efficacy of forming new memories (“learning”) vanishes drastically for many of us, as we grow older. This age-induced memory impairment (AMI) arose as one of the top public health threats during the past century. The understanding of this process eventually aims at the delay and/or prevention of such agerelated pathologies directly at the level of neurons. Neurons require efficient protein homeostasis (proteostasis) mechanisms to meet their, in general, high metabolic demands and to cope with stress to maintain cellular homeostasis. Loss of proteostasis is a ubiquitous hallmark of aging and disease (“neurodegeneration”), characterized by build-up of protein aggregates and organelle deterioration. Autophagy in turn has been found to be critical for organelle and protein homeostasis, with genetically provoked inhibition of autophagy causing neurodegeneration in autophagy-deficient mice and flies. Notably, autophagic clearance declines with progressing age, evident in a progressive accumulation of SQSTM1/p62/ref(2)p-positive aggregates. Conversely, genetic or pharmacological induction of autophagy by spermidine (an endogenous polyamine that induces autophagy) or rapamycin (an inhibitor of MTOR signaling, a pathway inhibiting autophagy) promotes clearance of aggregated proteins and likely as a direct consequence longevity/life span in several species.
The first kiss, the smell of your family’s cooking or a child’s first word: memories such as these are imprinted tightly into our brains and can be recollected even years later. However, the efficacy of forming new memories (“learning”) vanishes drastically for many of us, as we grow older. This age-induced memory impairment (AMI) arose as one of the top public health threats during the past century. The understanding of this process eventually aims at the delay and/or prevention of such agerelated pathologies directly at the level of neurons. Neurons require efficient protein homeostasis (proteostasis) mechanisms to meet their, in general, high metabolic demands and to cope with stress to maintain cellular homeostasis. Loss of proteostasis is a ubiquitous hallmark of aging and disease (“neurodegeneration”), characterized by build-up of protein aggregates and organelle deterioration. Autophagy in turn has been found to be critical for organelle and protein homeostasis, with genetically provoked inhibition of autophagy causing neurodegeneration in autophagy-deficient mice and flies. Notably, autophagic clearance declines with progressing age, evident in a progressive accumulation of SQSTM1/p62/ref(2)p-positive aggregates. Conversely, genetic or pharmacological induction of autophagy by spermidine (an endogenous polyamine that induces autophagy) or rapamycin (an inhibitor of MTOR signaling, a pathway inhibiting autophagy) promotes clearance of aggregated proteins and likely as a direct consequence longevity/life span in several species.
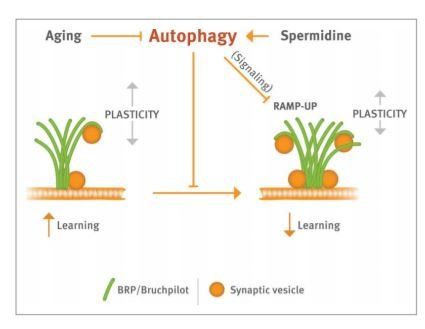
Previous work by the Sigrist and Madeo labs has shown that spermidine levels decline in aged Drosophila heads, similar to mice and humans, and that elevation of spermidine by simple feeding is sufficient to protect aged flies from AMI. These protective effects of spermidine feeding on learning are autophagy dependent, as already the loss of a single Atg7 gene copy effectively occludes spermidine effects on learning abilities. One challenge remaining was to understand how protecting learning abilities is mechanistically executed in neurons. Neurons release synaptic vesicles (SVs) filled with neurotransmitter at small cell-cell junctions, called synapses, to communicate with their post-synaptic neuron partners. Changes in the structural layout, and thus the release function of synapses (“synaptic plasticity”), is considered to be the key mechanism for learning, in the fruit fly and elsewhere. Reflecting their fundamental importance, the site of SV release, also called the active zone (AZ), is a highly specialized protein architecture patterned by a conserved scaffold protein called brp/bruchpilot, which by sequestering critical SV release factors such as unc-13 directly controls synaptic plasticity. A follow-up study recently published in PLoS Biology now directly makes a connection between spermidine treatment, autophagic control and synaptic plasticity. Somewhat contrary to expectations, this work found that AZs scaled up their size, provoking an increase in SV release at the synapses sensing odors in olfactory conditioning.
These age-dependent changes at AZs (termed RAMP-UP), were absent in aged animals fed with spermidine. Genetically mimicking RAMP-UP (flies with 4 instead of the normal 2 Brp gene copies) in fact provoked “early AMI," meaning that learning was already inhibited in young animals. Concerning neuronal information processing and learning, RAMP-UP might drive synapses toward the upper limit of their operational range, and thus saturate plasticity processes and consequently interfere with efficient learning. In other words, aging might undermine a homeostatic regime ensuring efficient synaptic plasticity and thus provoke a dwindling of cognitive performance. How exactly, however, would autophagic regulation come into play here? Gupta et al. (2016), found autophagic regulation is part of this homeostatic system, as atg7 null mutant animals show a RAMP-UP at a young age, mimicking the aging phenotype. Early RAMP-UP in atg7 mutants looks identical to RAMPUP in aged but genetically normal animals: presynaptic AZ proteins are elevated, SV proteins are also elevated but to a lower extent, while “postsynaptic” proteins forming the opposite side of the synaptic cell-cell junction are not affected by the RAMP-UP process. Moreover, spermidinemediated protection from RAMP-UP is occluded by the atg7mutant background, further strengthening the importance of autophagy for spermidine-mediated effects. Are AZ proteins (and synaptic vesicle proteins) direct targets of autophagy? The role of autophagy for turnover of synaptic proteins, unfortunately, is hardly investigated. That said, the chaperone protein Hsc70-4 was recently implicated to promote turnover of synaptic proteins bearing the specific-recognition motif “KFERQ," through endosomal microautophagy in Drosophila.
Interestingly, the levels of unc-13 are directly coupled to Brp, and unc-13 is also enriched in the RAMP-UP scenario of aged fruit flies. There is thus the possibility that the AZ proteins, whose levels increase with age (brp, Rbp, unc-13), might be direct substrates of “synaptic microautophagy." It should be interesting to further investigate the potential protective role of microautophagy, regulated via Hsc70-4, at synapses in suppressing the RAMP-UP. While macro- and microautophagy might directly control the age-induced synaptic specializations, other proteins and organelles with a role in aging might become relevant for the role of autophagy toward synaptic plasticity. For example, quality control of mitochondria (through mitophagy) might change the Ca2C signaling at synapses, a process of central importance for synaptic plasticity. Moreover, transport of autophagosomes might intersect with transport processes helping in the turnover of synaptic proteins. Finally, cell nonautonomous roles of autophagy for synaptic homeostasis, e.g. mediated by neuronal peptides known to potentially intersect with plasticity, cannot be excluded at this point in time. In fact, metabolic reprogramming of autophagy within neurons of the hypothalamus influences their neuropeptide secretion and thereby behavior, potentially operating at synapses as well. The past few decades have seen induction of autophagy as a reasonable strategy to help neurons to clear abnormal aggregates and survive. This study emphasizes spermidine as a natural modulator of autophagy to either slow down or prevent progression of neurodegenerative diseases with age, which are incurable so far.
The data presented in Gupta et al., 2016 strongly puts forth the idea that spermidine is capable of resetting the adult brain to a juvenile brain by acting at the level of the synapse itself, thus antagonizing age-induced cognitive disabilities. Figure 1 summarizes the suggestion that spermidine-triggered restoration of autophagy protects synapses from age-induced changes, and thus delays the normally occurring decline of memory formation. Given that spermidine is a physiologic, easy administrable substance, future research may consider its supplementation to counter age-dependent dementia.